Five Considerations for Non-U.S. MedTech Companies Entering the U.S. Market.

I recently participated in a panel focused on investment considerations for companies based outside the US. The questions from founders were consistent with what we hear in client engagements: the FDA pathway is understood in outline, but the operational details that determine timeline and capital efficiency are often misjudged. I believe there is a broader set of factors that drive what we call Translational Velocity™ — the speed at which a technology moves from concept to US clinical adoption. Below are the considerations we find most important.
Regulatory strategy belongs at the concept stage

The question we received on the panel — how early should founders think about compliance and regulatory strategy — has a short answer: at concept.
Pathway determination (510(k), De Novo, PMA), predicate device identification, and indications for use shape design decisions, testing protocols, and the clinical evidence plan. Companies that defer these decisions until after OUS commercialization often discover that data generated for other markets requires supplementation or repetition for a US submission. The pathways differ materially in time and capital: a 510(k) typically runs about 31 months and $3-5MM from concept to clearance, while De Novo (66 months, ~$5-15MM) and PMA (69 months) carry longer horizons. A Pre-Submission (Pre-Sub) through the Q-Submission program — which returns written FDA feedback within roughly 70 days — is a low-cost way to surface FDA expectations before they become rework.
OUS clinical data is usable — with conditions
Among the more common misunderstandings we see from non-US startups is the assumption that clinical data generated outside the US either transfers automatically or must be discarded entirely.
Neither is accurate. FDA accepts OUS data, but US generalizability must be addressed in the clinical trial population. Sponsors should be prepared to justify how the study population, standard of care, and clinical practices in the OUS setting represent the intended US use environment. Where gaps exist, a bridging strategy — rather than a full repeat — may be sufficient. This is a question worth raising in a Pre-Sub rather than discovering in a deficiency letter.
Foreign human factors data faces specific scrutiny
Human factors (HF) testing deserves separate attention because the acceptability bar differs from bench or clinical data.
In our experience, FDA has asked sponsors to justify how OUS HF testing represents US clinical users — including demographics,and language, education, health practices, and clinical terminology. FDA has also noted where OUS HF reports did not follow FDA's human factors guidance and requested justification for deviations. Practical implications: conduct HF studies against FDA's guidance framework from the outset, ensure complete English translation of all documentation (testing reports, device description, labeling), and budget for a US-based validation study where user populations differ meaningfully.
FDA programs can change the calculus — review all of them
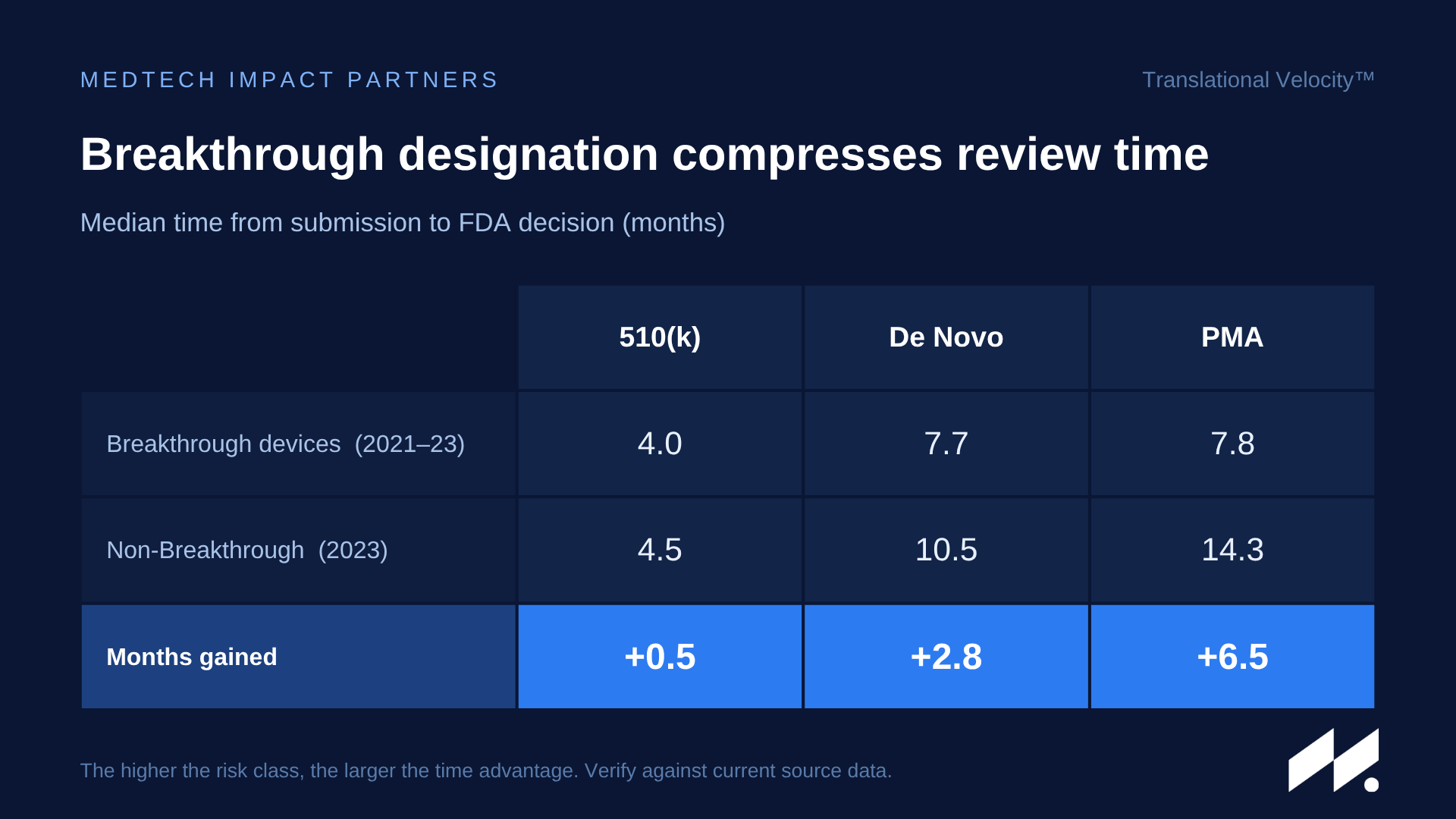
Non-US companies often default to the standard pathway without a structured review of FDA's program options.
Breakthrough Device Designation (BDD) can provide interactive review and prioritized engagement — and the timeline effect is measurable: Breakthrough-designated devices have reached decisions a median of 2.8 months faster via De Novo and 6.5 months faster via PMA than non-Breakthrough devices.
The Q-Submission program structures early dialogue. More recently, the TEMPO pilot (Technology-Enabled Meaningful Patient Outcomes), launched by CDRH in connection with the CMMI ACCESS model, offers certain digital health device manufacturers a route to supervised real-world use and evidence generation in chronic disease management — a development worth evaluating for SaMD and digital health companies weighing US entry. On the reimbursement side, CMS's proposed RAPID coverage pathway (April 2026) would align national coverage determinations with FDA authorization for certain Breakthrough devices, a reminder that market access planning extends beyond clearance: FDA authorization confirms safety and effectiveness, but coverage has historically taken years longer. A robust review of these programs at the strategy stage can shorten timelines and, in some cases, strengthen the reimbursement story.
Fundraising in MedTech runs on milestones — plan for slip
Capital in MedTech is tied to regulatory and clinical milestones in a way that differs from SaaS or AI.
The economic logic is concrete: written FDA feedback via Pre-Sub is associated with a post-seed value increase in the range of $500K–$1M, 510(k) clearance with $2M–$5M, clinical trial completion with $5M–$40M depending on pathway, and De Novo granting or PMA approval with increases approaching $50M. We encourage founders to structure raises around these milestones and mini-milestones — a Pre-Submission completed, a testing protocol agreed, a submission type accepted — rather than broad development phases.
Schedule slip is common, so raise size should reflect it: in the current environment, rounds in the $2M–$5M range sized to reach a defined regulatory milestone, with buffer, tend to position companies better than minimal raises. One additional consideration for founders raising in the next 12 months: non-dilutive sources are worth a structured look. ARPA-H, for example, funds in $2M–$20M increments that can support clinical data collection and FDA feedback, and where government matching programs are available, raising roughly $1M more alongside a match can extend runway without proportional dilution. Investors also weigh OUS revenue and milestones differently than US ones, so founders should be prepared to translate OUS traction into terms US investors recognize.
A closing consideration
For companies based outside the US, the practical step is a structured US regulatory gap assessment before the next raise — covering pathway, data acceptability, HF requirements, and program eligibility. The cost of the assessment is modest relative to the cost of repeating a study.
About the Author

Kwame Ulmer is Managing Partner at MedTech Impact Partners, a medical device regulatory strategy consulting firm.
He brings more than 20 years of experience evaluating medical technologies, including 12 years at the US Food and Drug Administration in progressive leadership roles such as Deputy Director and Branch Chief, and service as Vice President of Regulatory Affairs and Quality Assurance at Implant Direct, a Danaher subsidiary. He has personally evaluated more than 1,000 medical technologies.
Kwame is also a venture partner at Wavemaker Three-Sixty Health and the founder of MedTech Color, a nonprofit focused on representation in the medical device industry.